In today’s meeting the presentation for tomorrow’s seminar was finalized.
We also made the first draft of our Gantt schedule and we will most likely revise it, especially now in the beginning when we are still researching the different options of how to solve the problems in this project. A screenshot of the Gantt schedule can be seen below.
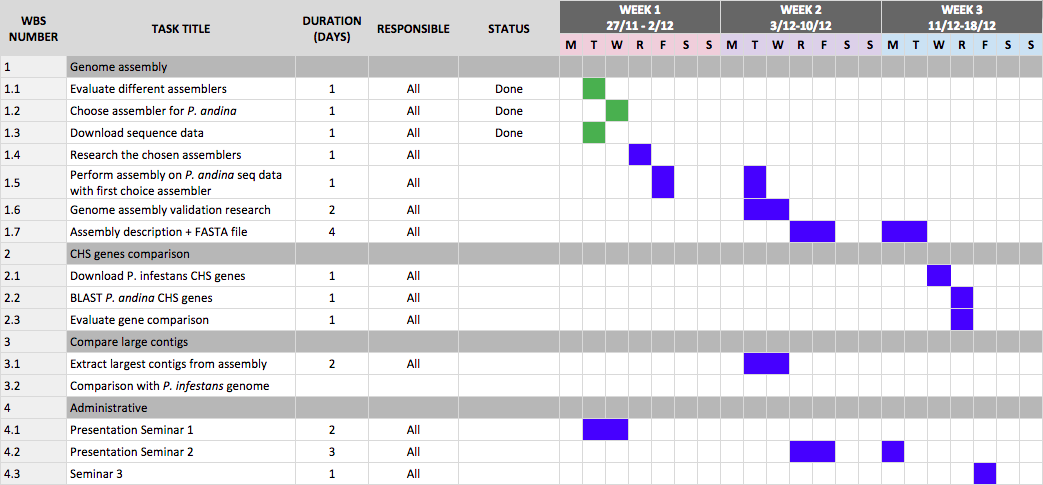
Current Gantt schedule
Choosing the assembler
One of the first discussions we had as a group was regarding the type of assembler we will use for our genome assembly. As mentioned in a previous post, a list of all assemblers available on UppMax was made (42 assemblers in total!). Out of these 42 assemblers, 20 assembler options could be removed due to different reasons:
- They are made for RNA-seq data specifically, and not DNA which is what we have (e.g. Trinity or Trans-ABySS),
- They are specifically designed for handling long-read sequencing data (e.g. FALCON or canu), from for example PacBio, but we have short reads from Illumina.
Out of the 22 other assemblers, 7 was removed due to either being made for analyzing metagenomics data (and not specifically for assembling genomes de novo), analyzing massively parallel sequencing data, or simply not being documented well enough for us to understand what they do at this point. An important factor is after all that the tool one uses not only is suited for the type of project, but also that it is well-documented and that people “out there” are using it. If the tool is well-used for similar projects, then that indicates it is a good tool (or that it is the only one available, but now we know that is not the case).
Left was then 15 assemblers that from quick research seemed to be good options. We decided today to look into the following assemblers in more detail:
- ABySS (there is a version that assembles genomes > 100 Mb)
- SOAPdenovo
- Velvet
In the next-coming days, we will look specifically into what input and output these assemblers have, if they have scaffolding as a part of the assembly, and generally how accurate they seem to be. Additionally, the actual documentation and syntax for how to use the assemblers will be looked into so that we can start the assembly process!
To summarize our biggest questions at this point:
- Once we have our first assembly, what are the best way of evaluating its accuracy?
- How do we best compare the results obtained from different assemblers?
- Why and how do we take out the largest contig from our genome of interest, and compare that to the reference genome (as instructed in the project)? Can one compare the whole genomes?