This is a long post as a lot have happened. Take your time!
Summary
Yesterday our group had a Skype meeting as we could not meet in person. We discussed what has been done since last time regarding the BLAST-part of the project, which is the following:
- All of us have used BLAST to annotate the CHS-genes against the assembled contigs using ABySS
- Panos has specifically annotated the P. infestans genes whereas me and Maria used all species’ CHS-genes.
- We have decided to continue with the dataset where a cut-off e-value of 0.05 was used (as allowing all e-values simply results in way too much data)
- The data have been sorted according to the order of p-identity, length and e-value
- We decided to first analyze the P. infestans data
- We decided that Maria continues with looking the BLAST results for all species we got genes for, Panos will update the assembly description and look into
MUMmer, and I will also look intoMUMmerand take out the largest contig from the P. andina assembly data.
BLAST results of P. infestans CHS-genes against assembled P. andina genome
This is the top 20 rows of the P. infestans data sorted as mentioned above.
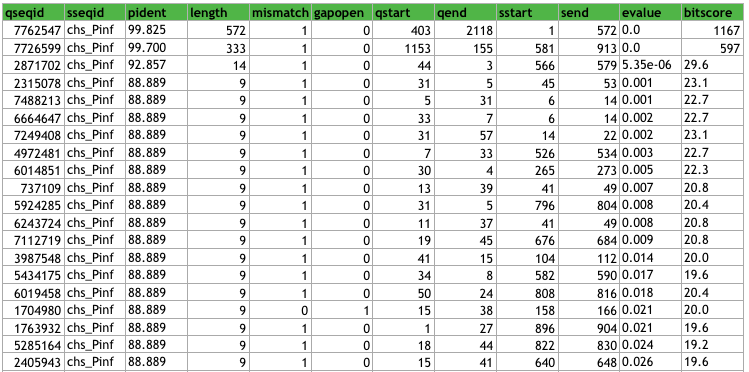
Top 20 hits after BLAST with P. infestans CHS-genes against assembled P. andina contigs.
When looking through the data, we can see that a lot of contigs overlaps (as expected), for example, the third hit (with length 14) represents a sequence that begins in the first hit (sstart = 3) and ends within the second hit (send = 579).
We can also conclude that the longest contig of the P. andina assembly in which a CHS-gene coding sequence from P. infestans can be found, is 572 bases long. However, note that this does not necessarily represent the longest contig.
BLAST evaluation
Here I will try to explain the parameters a bit and my view on them with regards to our results.
E-value
As mentioned, the parameters we look at are E-value, length that has aligned, and the identity. The E-value gives us information about the likelihood that the given sequence, i.e. the CHS-gene sequence, match purely by chance among our contigs. The lower the E-value, the less likely it is that the match was a result of a random chance, and the more significant it is.
For our two hits, the E-value is 0. To be honest, I initially thought the E-value approaches zero, but that it never can actually be 0, as this would mean there is no chance what so ever that it occurred by chance. I am not entirely sure why that can be at the moment, as there is “always” a chance (although infinitely small). Perhaps BLAST outputs 0 when the value is small enough, I will read into this more when I have time.
Anyway, what we can conclude with such E-values is that there is an extremely high confidence that the match is a result of a homologous relationship between P. andina and P. infestans for the CHS-gene, which is also expected as we knew beforehand that P. infestans was the closest relative to P. andina. It should also be mentioned that this is one of those genes that one can expect to be conserved over time between the species as it is vital for building up the cell walls, and thus we could expect to see these genes (and consequently chitin synthases) in most species with these type of cell wall structures and related phenotypes.
Identity and length
I think these two parameters are quite straight-forward, and the identity is very high for both hits, i.e. the extent to which the CHS-gene sequence have the same residues (and nucleotides) at the same positions is very high. For contig 7762547, it is 99.825%, and for contig 7726599 it is 99.700. For the contig 2871702, the identity is 92.657% which is still very high, but the length is very small and this contig is not really interesting for further analysis compared to one being almost 600 nt, although finding a much longer contig is ofcourse of interest for us.
Confirming contig sequence
Just to test the contig sequence from the P. andina assembly that matched against the P. infestans CHS-gene sequence, we used the webbased BLASTn tool for the contig sequence against everything for all three hits 7762547, 7726599, and 2871702. The results can be seen in the three figures below.
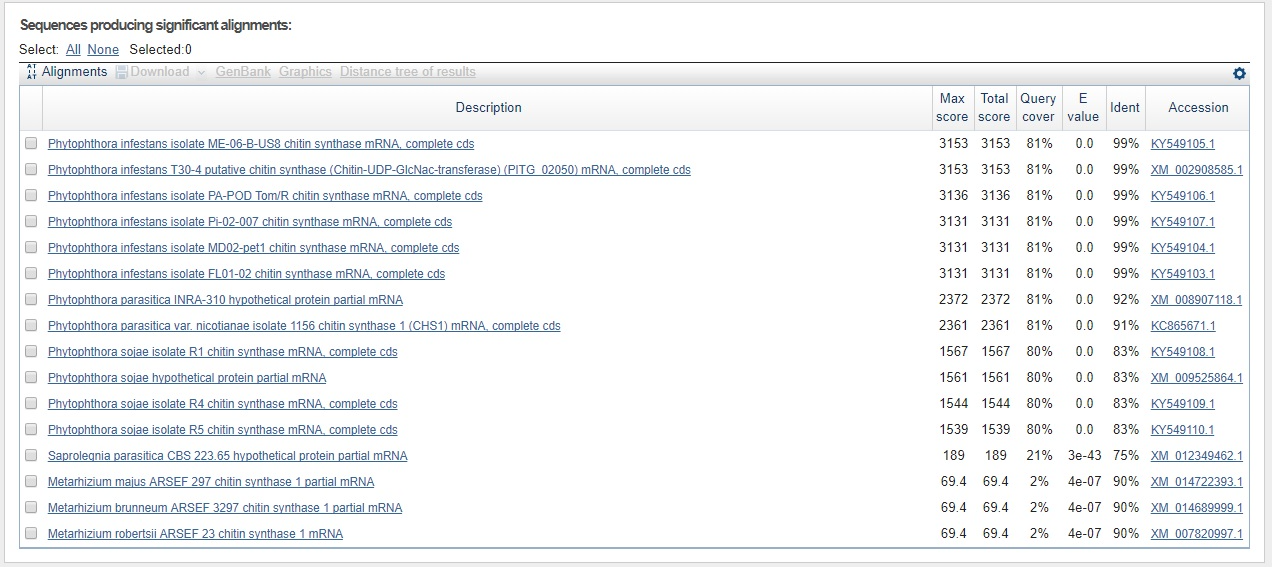
Longest contig sequence (572 nt) for P. infestans BLASTed against all with blastn.
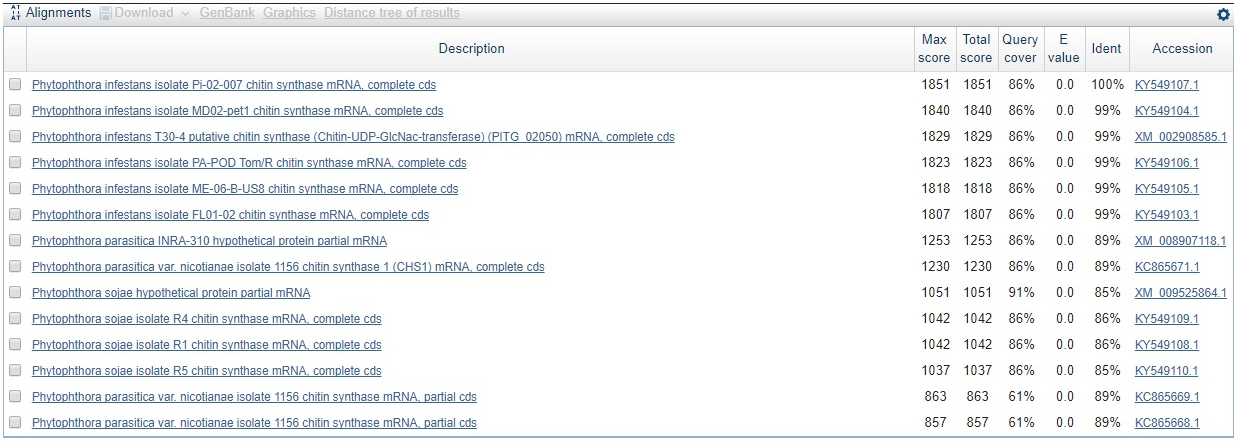
Second longest (333 nt) contig sequence for P. infestans BLASTed against all with blastn.
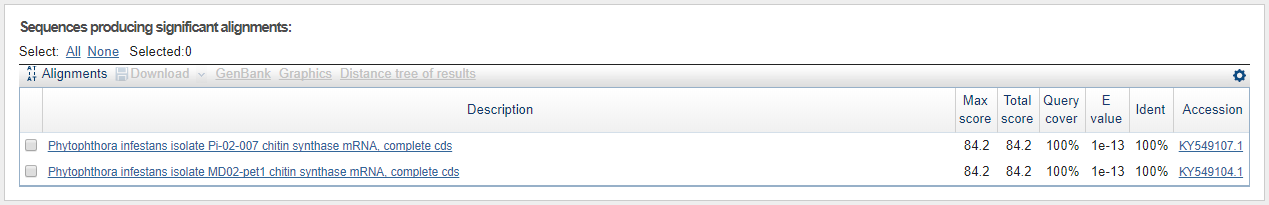
Shortest contig sequence (14 nt) worth looking into (based on the statistics parameters)for P. infestans BLASTed against all with blastn.
Not only have we confirmed that the sequence is indeed that of CHS-genes from P. infestans, but we can already here see that this sequence is found in other species as well. Perhaps most interesting is that the sequence is found with the same query cover and E-value, and just a slightly lower identity than that for P. infestans, in a species called Phytophtora parasitica which was not in our list of genes. This is also seen for the second hit which makes sense since the contig is part of the same gene sequence as seen from the sequence start- and end values, and knowing that the coding part of the P. infestans CHS-gene is around 2600 bp in total.
Longest contig for sequence comparison
To investigate the similarity between P. andina and P. infestans further, we will use MUMmer to compare the contigs against a genome of P. infestans that was published in 2009. To do this we need to take out the largest contig which is currently my responsibility of the project.
The .fa-output file from ABySS looks like this (10 first contig sequences):
>0 23 482
GCTGATCCTCGTGACTGTCGGCT
>1 23 79
CCTGACGAGCAGATCGGAAGAGC
>3 27 259
TCTATCGTCGAGGATGATCTCGACTTC
>4 23 11
ATCGCAATTTGAGCCACTCATAG
>5 23 5876
CTTTGGTGGTTTAACATTGTTTC
>7 23 29
TGAAGCTGCATTTGCTCTTGGTG
>8 24 86
GATCTATTGTATCACGTTCGCCAT
>9 26 565
AAGATATGTGTATCGAAAGAACCGTG
>11 24 9
CTTTGGTGGTTTAACATTGTTTTC
>12 26 271
ATAACGTGCAGCATCTTCAGGATGAA
For each sequence, identified with >, there are three numbers. The first one is the sequence id, the second is the number of bases, and the third is the number oh k-mers that were mapped to the sequence during assembly. Below is how the largest contig was obtained.
A regex helper function with gawk was defined to be able to take out a specific group.
$ function regex1 { gawk 'match($0,/'$1'/, ary) {print ary['${2:-'1'}']}';
Then, using regex, the sequence identifier (for example >12 26 271) was matched and the length of the sequences was sorted numerically.
$ cat assembled-unitigs.fa | regex1 '>[0-9]+\s([0-9]+).*' | sort -n > sorted-sizes.txt
The largest contig is found at the end of the output file sorted-sizes.txt.
$ tail sorted-sizes.txt
8584
8732
9157
9484
9690
9951
10148
10355
11835
15812
The largest contig is 15812 nt. This contig will be used in the next step to compare P. andina with P. infestans.
Planning ahead
Tomorrow, the 15th of December, we will have a meeting and go through the BLAST results as well as try and test MUMmerfor the first time. We will try and finalize the assembly description and the BLAST results as a whole, as well as discuss what our results mean for the project and the research in general. I am also glad that we are on schedule with everything that we have planned, and as it looks now, most of the work should be done in the middle of next week.