After the presentation on Seminar 1, our group sat down to discuss our first practical approach to this project - the assembly of the P. andina genome. As mentioned in the previous post, we dug into the our “top three” assemblers based on what we had found so far. I specifically looked for more information about the SOAPdenovo assembler which seems like a good option in terms of being designed for Illumina short-reads and assembly of human-sized genomes. As a conclusion from having read some articles and general documentation, as well as forum posts regarding the topic, SOAPdenovo works well on fungi genomes and accepts fastq as input format which is what we are working with. However, as more information was found on ABySS, we decided to start with this assembler as our first approach. Another advantage with using this one now is because two other groups are using it too, and perhaps we can exchange some tips!
When talking to Lars Arvestad after the seminar, we were told that our data is not “the best data”, and that the assembly might not be fantastic. We looked at the data (see figure below) and could indeed see that it is single end-reads (while most assembler mention using paired-end or/and mate-pair reads).
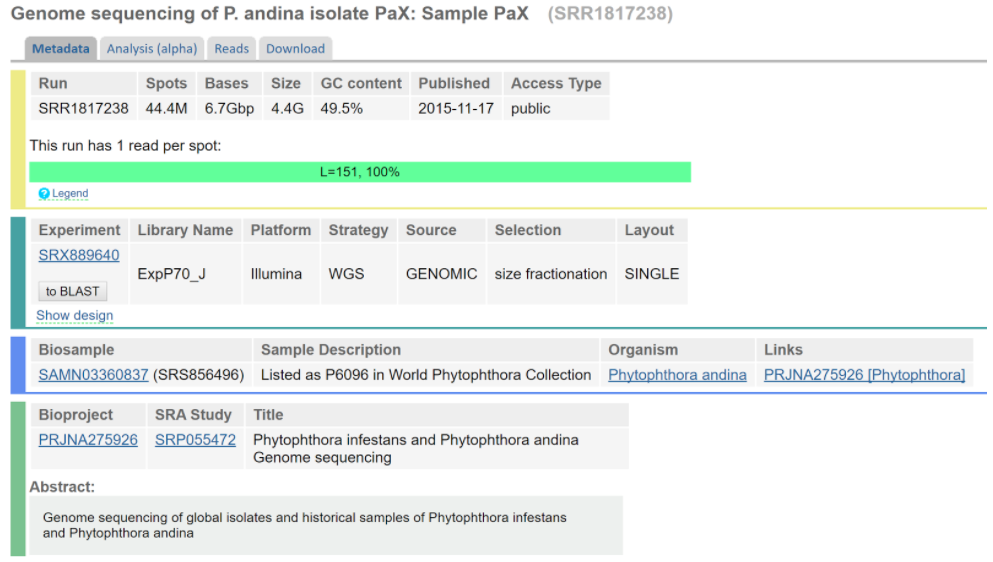
Screenshot of the sequencing data web page.
We are not sure yet how this will affect our assembly, and my thinking is that we will have to experiment a bit with the options available for ABySS and see what gives a better or worse assembly. For example, we could experiment with different k-mer sizes. However, I think we need to decide whether if it is more important to have an assembly that covers most of the genome, or to have a very accurate assembly of a part of the genome. From what I have understood it, it will be a bit of a trade-off.
The data
Today I also downloaded the sequencing data, both the reads and the CHS-gene files, to my folder on Rackham (UppMax). However, the format of the sequencing data file was in .gz-format, and I could not find a way to unzip that file on Rackham. Therefore I downloaded it locally to my computer, extracted it, and uploaded the acquired .fastq-file back to my folder on Rackham. This took a while considering the size of the files, but now we could take a look that the content which was as expected. Below is an example of a read.
@SRR1817238.2 HWI-ST897:215:H0RT3ADXX:1:1101:1174:2066/1
ATGAAAGTCCGGAGGATTCGGAGGAGGCTCGAGCCGTTGATGACGCCTTGGAGGAGGACACACGACGGGCCTTGTCCCAGTCCCGATCTGAAGCCCGGCGA
AGATCCTTTCAACCCCGCCTCCACAAGTCGCTGGTACCGCCGGATCCGGG
+
:?BABDDFFFCF@FH<CGFGIEHIGGHFHDHDFEIFHIJCGHHHHDFFDEEAED=@D8?C?CB=BBDDDBDDDDD@@CDCD@CC<5>@BCACCCBD<@9@>
9>BCCCCCDCDDDDDDB@DBDB?CDDBDDDDDDB?CDCBDBBDDDBD<@#
Using the following command (and assuming there is not subsequent score of @SRR) the following total of number of reads can be counted:
grep -o '@SRR' SRR1817238.fastq | wc -l
44445500
Regarding the CHS-genes, I seems like we do not have permission to copy that file from where it is deposited at UppMax (/proj/g2017020/nobackup/Phytophthora_andina) to our own home directory. This means we can not view the data at the moment, but we will still be able to use it for example with BLAST directly on Rackham.
That’s it for now. Tomorrow we will try and assemble the genome.